1.2 Pharmacokinetics and Pharmacodynamics Introduction
Learning Outcomes
- Apply understanding of the routes of administration and related nursing considerations.
- Correlate the pharmacology terms with their importance to medication safety.
- Articulate the four stages of pharmacokinetics in relation to nursing practice: absorption, distribution, metabolism, and excretion.
- Apply the principles of pharmacokinetics including first-pass effect, protein binding, onset/peak/duration, therapeutic range, and lipid and water solubility.
- Discuss and apply pharmacokinetic principles to life span considerations.
- Explain the various drug forms and identify the advantages and disadvantages of the dose forms and drug delivery.
Introduction
A nurse is preparing to administer some medications to a client, with the overall goal to achieve a desired effect. For a medication to reach the target cells to have this desired effect, it must first be given in sufficient quantities, produce the needed physiological changes, overcome any barriers that impede the medications effects, and then eventually be excreted from the body. This process is called pharmacokinetics, which is the study of what the body does to the drug. It involves the processes of absorption, distribution, metabolism, and excretion of drugs. Pharmacokinetics is probably the most important concept to understand within the study of pharmacology and will be the focus of the first part of this chapter.
Why does a medication work differently for different people? Some medications will have a dramatic effect that may include adverse reactions, while for other people, it may have the intended effect or maybe even a subtherapeutic response. These drug-induced changes in a client’s physiological function and the differences in how a client responds to medications is the study of pharmacodynamics. Pharmacodynamics is defined as the study of what a drug does to the body or the biological response to the drug. Pharmacodynamics will be the focus of the second part of this chapter.
The Relationship Between Pharmacokinetics and Pharmacodynamics
Pharmacokinetics and pharmacodynamics both need to be considered when administering medications. Both processes are closely related but distinctly different. Practitioners need to consider both processes when prescribing a medication to be able to make adjustments based on physiological responses and client factors (Grogan & Preuss, 2023). Nurses also must consider both processes before administering a medication; this helps them decide the best route to administer it, monitor the patient for adverse and intended effects, and evaluate the medication’s overall effectiveness.
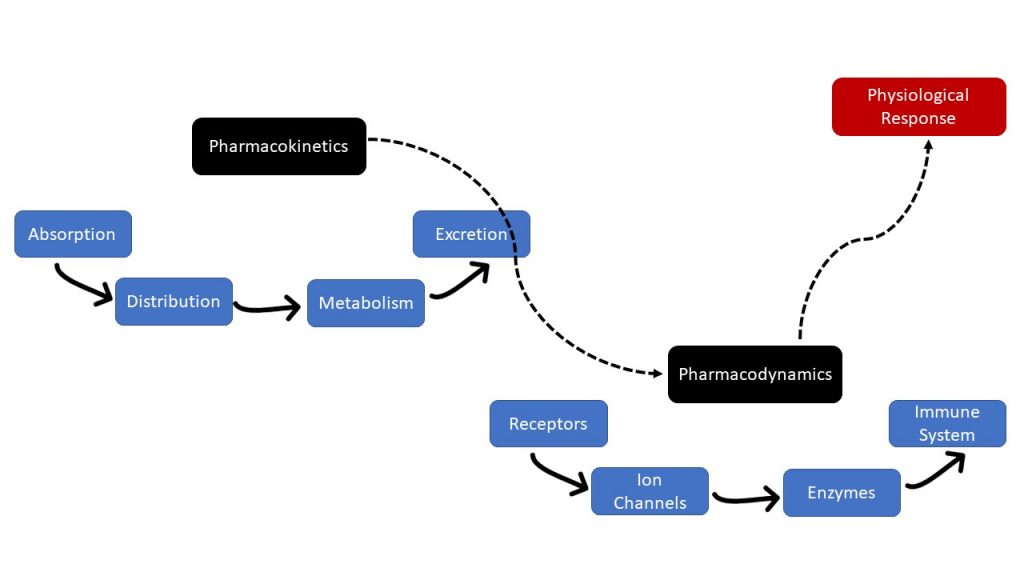
Figure 1.2.1 displays the relationship between pharmacokinetics and pharmacodynamics.
Pharmacology Principles — The Movement of Drugs Through Cell Membranes
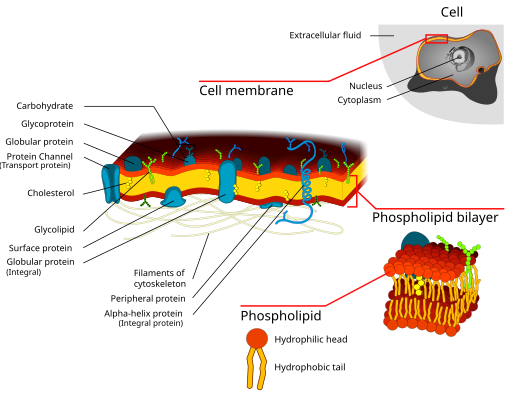
For drugs to work, they need to cross cell membranes. All phases of pharmacokinetics involve drug movement across membranes. Drugs must penetrate these membranes to produce their effects. For example, drugs cross a membrane to enter the bloodstream from the administration site, to leave the vascular system to enter the target site, and to be excreted from the body (Burcham & Rosenthal, 2019).
Recall that cell membranes are biologic barriers composed of layers of individual cells. They selectively inhibit the passage of molecules. These layers of individual cells are very close to one another. So close that most drugs must pass through the cells instead of between the cells. Furthermore, a cell membrane is composed of a bimolecular lipid matrix that determines the membrane permeability. There are hydrophilic heads on the outside with lipophilic chains between these heads, creating a sandwich effect. Some cell membranes are slightly modified to allow more specialized functions. For example, in the renal glomerular endothelium, gaps exist between cells to allow for large molecules to pass through as part of filtration. This will be discussed in more detail in the distribution section.
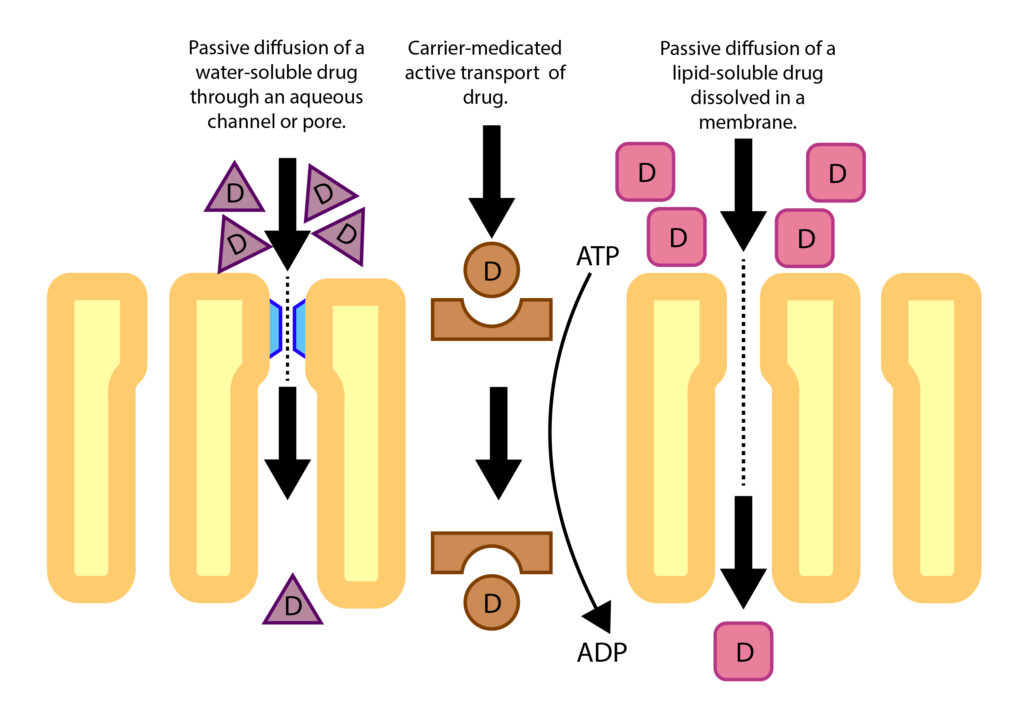
For a drug to pass through a membrane, it can do so in two different ways: passive transport or active transport.
Passive transport implies that no energy is required for the molecule to move through the cell membrane. There are a few ways drug molecules can move by passive transport:
- Diffusion: Drug molecules pass through aqueous channels or pores. These channels or pores are very small and are specific to certain molecules. Water-soluble drug molecules and small molecules can move through the pore along with water. Substances that can pass through a channel are potassium, sodium, and water-soluble drugs, such as acetaminophen.
- Passive Diffusion (most common): Drug molecules directly penetrate the membrane. Most drugs are large molecules that cannot pass through a channel or pore and do not have a transport system to help them cross cell membranes. Passive diffusion is a movement from a high concentration area to a lower concentration one. Many drugs are weak bases or weak acids and exist in either an ionized or non-ionized form, depending on the pH. A non-ionized drug is lipid soluble and will diffuse easily through the cell membrane. With the old rule of “like dissolves like,” since the cell membrane is primarily composed of lipids, a drug that is lipid soluble (lipophilic) will directly penetrate membranes. An example of lipid-soluble drugs are opiates, such as fentanyl.
- Facilitated Passive Diffusion: Drug molecules combine with a carrier protein to cross the cell membrane. The rate of diffusion of the drug molecule-carrier down a concentration gradient is faster than just diffusion alone. An example is a glucose molecule.
Diffusion rates are also influenced by a number of factors such as molecular size, concentration gradient, ionization, and lipid solubility. Recall that drugs can be hydrophilic (polar molecules and ions), meaning they cannot directly penetrate membranes, or lipophilic, meaning they can penetrate membranes due to the lipid bilayer. Drugs that are lipophilic are readily absorbed after administration and can easily reach target sites. We will discuss these concepts later in this chapter.
Active transport of molecules requires energy to move along the gradient:
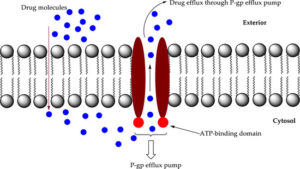
- Passage With the Aid of a Transport System: A transport system or carrier can move drugs from one side of a cell membrane to the other side. All transport systems are selective (will only carry certain drugs or molecules) and need energy. Some drugs use a transport system but are limited to drugs that are structurally similar to endogenous substances, such as vitamins or amino acids. An example of a drug using active transport is levodopa for Parkinson’s Disease. In the kidney, renal excretion would be very slow if not for a transport system that pumps the drugs from the blood to the renal tubules. P-glycoprotein (PGP), also referred to as a multidrug transporter protein, is the most commonly known protein that transports drugs out of cells (efflux pump). PGP is located at many sites, including the liver, kidney, placenta, intestines, and in the capillaries of the brain.
P-glycoprotein (PGP) is an example of a multi-drug transporter that transports drugs out of cells. It is a protein that is present in many cell membranes, including in the liver, kidneys, placenta, intestines, and brain capillaries. For example, it transports drugs across the membrane from the blood to the intestinal lumen, reducing drug absorption into the blood.
Lipid-Soluble Drugs Versus Water-Soluble Drugs
Drugs can be lipid (lipophilic) soluble or water soluble (hydrophilic), although most drugs are lipophilic.
Drugs that are lipid soluble:
- readily diffuse across cell membranes (faster absorption),
- pass the blood brain barrier more easily,
- are stored in fat tissue, and
- have a higher lipid solubility (i.e., are more readily absorbed than those with low lipid solubility).
Drugs that are water soluble:
- require a transport system to penetrate the cell membrane or move through a channel,
- stay in the bloodstream longer but are not stored in fat tissue, like lipophilic drugs,
- typically easily metabolized through hydrolysis,
- undergo renal excretion faster, and
- often have fewer side effects, depending on the distribution and clearance.
What are examples of water-soluble drugs? Some vitamins are water soluble. Vitamin B and C are two examples. They are not stored in fat tissue but, instead, stay in the bloodstream or in the interstitial tissues nearby. They are also more rapidly excreted from the body.
Other hydrophilic drugs include the antibiotic gentamycin and the beta blocker atenolol.
What are examples of lipid-soluble drugs? Many drugs are lipid soluble, including phenytoin and diazepam. For vitamins, some lipid-soluble vitamins include A, D, E, and K. If taken in excess, they are stored in fat tissue.
Drugs have varying degrees of lipid solubility, with some highly lipid soluble. For example, let us compare two opioids, morphine and fentanyl. Both are lipid soluble, but fentanyl is highly lipid soluble. Consider what this may mean for the onset and duration of action.
Significance to practice: You may wonder why you need to understand drug movement and cell membranes. Understanding these basic principles will help you apply the concepts of pharmacokinetics and pharmacodynamics when administering medications and monitoring clients for their effects.
References
Burcham, J. R., & Rosenthal, L. (2019). Lehne’s pharmacology for nursing care (10th ed.). Elsevier.
Chippewa Valley Technical College, Egert, A., Lee, K., & Gill, M. (2023). Fundamentals of nursing pharmacology (1st Canadian ed.). BCcampus. https://opentextbc.ca/nursingpharmacology/chapter/1-9-examining-effect/
Grogan, S., & Preuss, C. V. (2023). Pharmacokinetics. Statpearls. Retrieved October 20, 2025, from https://www.ncbi.nlm.nih.gov/books/NBK557744/
Le, J. (2024). Drug absorption. MSD Manual Professional Version. https://www.msdmanuals.com/professional/clinical-pharmacology/pharmacokinetics/drug-absorption
Stielow, M., Witczyńska, A., Kubryń, N., Fijałkowski, Ł., Nowaczyk, J., & Nowaczyk, A. (2023). The bioavailability of drugs—The current state of knowledge. Molecules, 28(24), Article 8038. https://doi.org/10.3390/molecules28248038
Media Attributions
- Figure 1.2.1 Figure 1.2b The Relationship Between Pharmacokinetics and Pharmacodynamics by Chippewa Valley Technical College et al. (2023), via BCcampus, is used under a CC BY 4.0 license.
- Figure 1.2.2 Cell membrane detailed diagram 3 by Dhatfield [a derivative work of “Cell_membrane_detailed_diagram.svg” by LadyofHats/Mariana Ruiz], via Wikimedia Commons, is used under a CC BY-SA 3.0 license.
- Figure 1.2.3 The Mechanism of Drug Import and Efflux Through P-gp Efflux Pum by Gandla et al. (2023), via Heliyon, is used under a CC BY-NC-ND 4.0 license.
- Figure 1.2.4 [may need info for combined images]
- Figure 1.2.5 Passive transport vs. active transport was created by TRU Open Press, adapted from Passive vs Active Membrane Transport by LSumi, via Wikimedia Commons, is used under a CC BY-SA 4.0 license.
Long Descriptions
Figure 1.2.1 Long Description:
- Pharmacokinetics
- Absorption
- Distribution
- Metabolism
- Excretion
- Pharmacodynamics
- Receptors
- Ion Channels
- Enzymes
- Immune System
- Physiological Response [Return to Figure 1.2.1]

{kind=link}
{kind=link}